
SCGV
 https://ascopubs.org/na101/home/literatum/publisher/asco/journals/content/cci/2020/cci.2020.4/cci.19.00171/20200519/images/large/cci.19.00171f3.jpeg
https://ascopubs.org/na101/home/literatum/publisher/asco/journals/content/cci/2020/cci.2020.4/cci.19.00171/20200519/images/large/cci.19.00171f3.jpeg
Methods
linear multiple view single scale single focus contiguous no abstraction linear parallel arrangement no interconnection segment contiguous type segment sparse type point contiguous type point sparse typeTool
| Access Format | programming library |
| Supported Files | other |
| License | Creative Commons Attribution 4.0 License |
| Tool name | SCGV |
| Tool Link | https://github.com/KrasnitzLab/SCGV |
| Documentation |
Paper
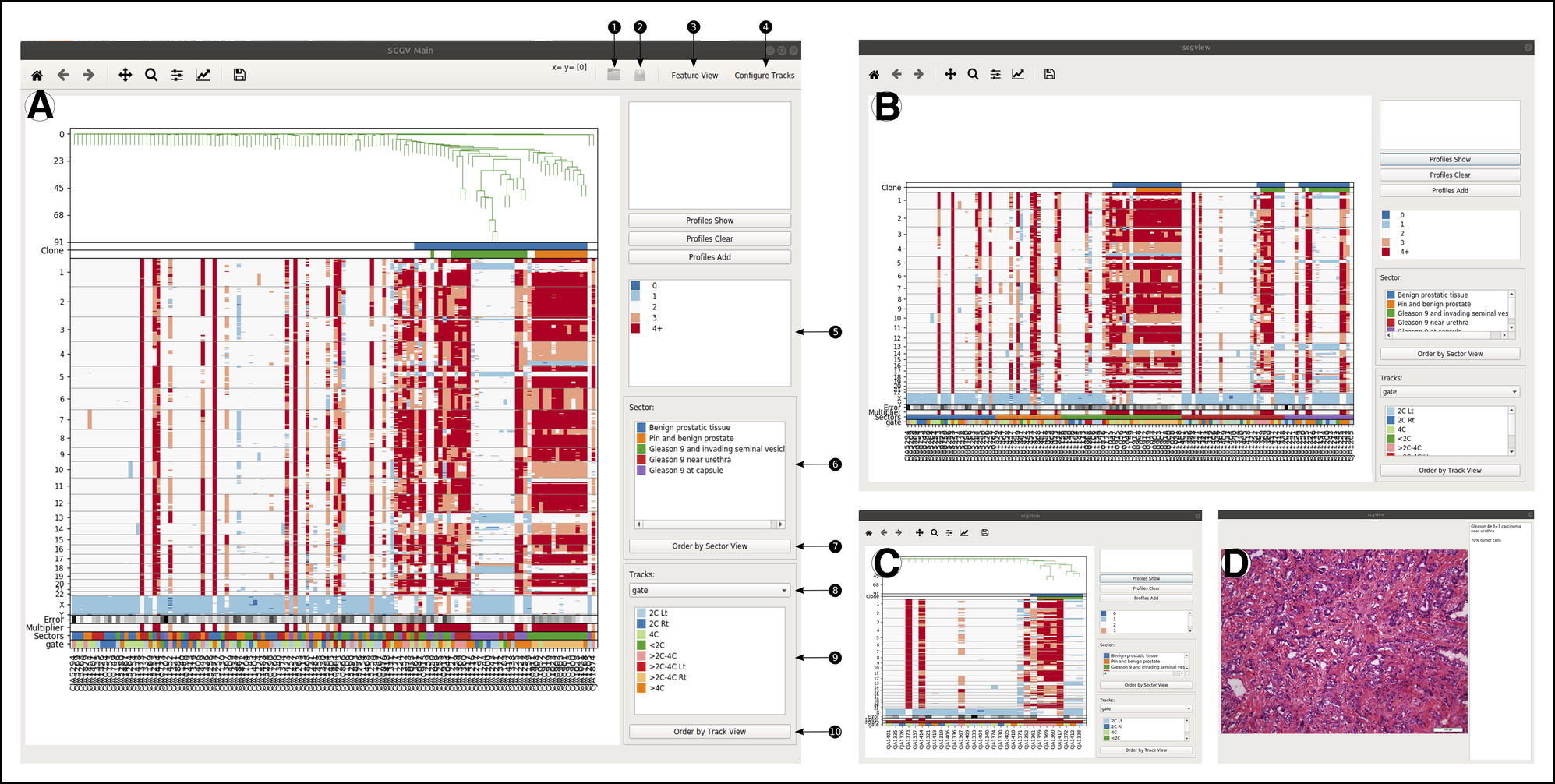
Integrated Computational Pipeline for Single-Cell Genomic Profiling
Chorbadjiev, Lubomir, et al. Integrated Computational Pipeline for Single-Cell Genomic Profiling. JCO Clinical Cancer Informatics 4 (2020): 464-471.
Abstract
Copy-number profiling of multiple individual cells from sparse sequencing may be used to reveal a detailed picture of genomic heterogeneity and clonal organization in a tissue biopsy specimen. We sought to provide a comprehensive computational pipeline for single-cell genomics, to facilitate adoption of this molecular technology for basic and translational research. The pipeline comprises software tools programmed in Python and in R and depends on Bowtie, HISAT2, Matplotlib, and Qt. It is installed and used with Anaconda. Here we describe a complete pipeline for sparse single-cell genomic data, encompassing all steps of single-nucleus DNA copy-number profiling, from raw sequence processing to clonal structure analysis and visualization. For the latter, a specialized graphical user interface termed the single-cell genome viewer (SCGV) is provided. With applications to cancer diagnostics in mind, the SCGV allows for zooming and linkage to the University of California at Santa Cruz Genome Browser from each of the multiple integrated views of single-cell copy-number profiles. The latter can be organized by clonal substructure or by any of the associated metadata such as anatomic location and histologic characterization. , The pipeline is available as open-source software for Linux and OS X. Its modular structure, extensive documentation, and ease of deployment using Anaconda facilitate its adoption by researchers and practitioners of single-cell genomics. With open-source availability and Massachusetts Institute of Technology licensing, it provides a basis for additional development by the cancer bioinformatics community.